|
|
@@ -23,20 +23,22 @@
|
|
|
% file structure/format <-> datatypes. länger beschreiben: e.g. File formats to store dna
|
|
|
% 3.2.1 raus
|
|
|
|
|
|
-
|
|
|
\section{Compression aproaches}
|
|
|
The process of compressing data serves the goal to generate an output that is smaller than its input data.\\
|
|
|
-In many cases, like in gene compressing, the compression is idealy lossless. This means it is possible for every compressed data, to receive the whole information, which were available in the origin data, by decompressing it.\\
|
|
|
+In many cases, like in gene compressing, the compression is ideally lossless. This means it is possible for every compressed data, to receive the whole information, which were available in the origin data, by decompressing it.\\
|
|
|
Before going on, the difference between information and data should be emphasized.\\
|
|
|
% excurs data vs information
|
|
|
-Data contians information. In digital data clear, physical limitations delimit what and how much of something can be stored. A bit can only store 0 or 1, eleven bits can store up to $2^11$ combinations of bits and a 1\acs{GB} drive can store no more than 1\acs{GB} data. Information on the other hand, is limited by the way how it is stored. In some cases the knowledge received in a earlier point in time must be considered too, but this can be neglected for reasons described in the subsection \ref{k4:dict}.\\
|
|
|
+Data contains information. In digital data clear, physical limitations delimit what and how much of something can be stored. A bit can only store 0 or 1, eleven bits can store up to $2^11$ combinations of bits and a 1 Gigabyte drive can store no more than 1 Gigabyte data. Information on the other hand, is limited by the way how it is stored. In some cases the knowledge received in a earlier point in time must be considered too, but this can be neglected for reasons described in the subsection \ref{k4:dict}.\\
|
|
|
% excurs information vs data
|
|
|
-The boundaries of information, when it comes to storing capabilities, can be illustrated by using the example mentioned above. A drive with the capacity of 1\acs{GB} could contain a book in form of images, where the content of each page is stored in a single image. Another, more ressourcefull way would be storing just the text of every page in \acs{UTF-16}. The information, the text would provide to a potential reader would not differ. Changing the text encoding to \acs{ASCII} and/or using compression techniques would reduce the required space even more, without loosing any information.\\
|
|
|
+The boundaries of information, when it comes to storing capabilities, can be illustrated by using the example mentioned above. A drive with the capacity of 1 Gigabyte could contain a book in form of images, where the content of each page is stored in a single image. Another, more resourceful way would be storing just the text of every page in \acs{UTF-16}. The information, the text would provide to a potential reader would not differ. Changing the text encoding to \acs{ASCII} and/or using compression techniques would reduce the required space even more, without loosing any information.\\
|
|
|
% excurs end
|
|
|
-In contrast to lossless compression, lossy compression might excludes parts of data in the compression process, in order to increase the compression rate. The excluded parts are typicaly not necessary to persist the origin information. This works with certain audio and pictures formats, and in network protocols \cite{cnet13}.
|
|
|
-For \acs{DNA} a lossless compression is needed. To be preceice a lossy compression is not possible, because there is no unnecessary data. Every nucleotide and its position is needed for the sequenced \acs{DNA} to be complete. For lossless compression two mayor approaches are known: the dictionary coding and the entropy coding. Methods from both fields, that aquired reputation, are described in detail below \cite{cc14, moffat20, moffat_arith, alok17}.\\
|
|
|
+In contrast to lossless compression, lossy compression might excludes parts of data in the compression process, in order to increase the compression rate. The excluded parts are typically not necessary to persist the origin information. This works with certain audio and pictures formats, and in network protocols \autocite{cnet13}.
|
|
|
+For \acs{DNA} a lossless compression is needed. To be precise a lossy compression is not possible, because there is no unnecessary data. Every nucleotide and its position is needed for the sequenced \acs{DNA} to be complete. For lossless compression two mayor approaches are known: the dictionary coding and the entropy coding. Methods from both fields, that aquired reputation, are described in detail below \autocite{cc14, moffat20, moffat_arith, alok17}.\\
|
|
|
|
|
|
\subsection{Dictionary coding}
|
|
|
+\textbf{Disclaimer}
|
|
|
+Unfortunally, known implementations like the ones out of LZ Family, do not use probabilities to compress and are therefore not in the main scope for this work. To strenghten the understanding of compression algortihms this section will remain. Also a hybrid implementation described later will use both dictionary and entropy coding.\\
|
|
|
+
|
|
|
\label{k4:dict}
|
|
|
Dictionary coding, as the name suggest, uses a dictionary to eliminate redundand occurences of strings. Strings are a chain of characters representing a full word or just a part of it. For a better understanding this should be illustrated by a short example:
|
|
|
% demo substrings
|
|
|
@@ -44,27 +46,30 @@ Looking at the string 'stationary' it might be smart to store 'station' and 'ary
|
|
|
% end demo
|
|
|
The dictionary should only store strings that occour in the input data. Also storing a dictionary in addition to the (compressed) input data, would be a waste of resources. Therefore the dicitonary is made out of the input data. Each first occourence is left uncompressed. Every occurence of a string, after the first one, points to its first occurence. Since this 'pointer' needs less space than the string it points to, a decrease in the size is created.\\
|
|
|
|
|
|
-
|
|
|
% unuseable due to the lack of probability
|
|
|
% - known algo
|
|
|
\subsubsection{The LZ Family}
|
|
|
The computer scientist Abraham Lempel and the electrical engineere Jacob Ziv created multiple algorithms that are based on dictionary coding. They can be recognized by the substring \texttt{LZ} in its name, like \texttt{LZ77 and LZ78} which are short for Lempel Ziv 1977 and 1978. The number at the end indictates when the algorithm was published. Today LZ78 is widely used in unix compression solutions like gzip and bz2. Those tools are also used in compressing \ac{DNA}.\\
|
|
|
-\ac{LZ77} basically works, by removing all repetition of a string or substring and replacing them with information where to find the first occurence and how long it is. Typically it is stored in two bytes, whereby more than one one byte can be used to point to the first occurence because usually less than one byte is required to store the length.\\
|
|
|
+\acs{LZ77} basically works, by removing all repetition of a string or substring and replacing them with information where to find the first occurence and how long it is. Typically it is stored in two bytes, whereby more than one one byte can be used to point to the first occurence because usually less than one byte is required to store the length.\\
|
|
|
% example
|
|
|
|
|
|
% (genomic squeeze <- official | inofficial -> GDC, GRS). Further \ac{ANS} or rANS ... TBD.
|
|
|
\ac{LZ77} basically works, by removing all repetition of a string or substring and replacing them with information where to find the first occurence and how long it is. Typically it is stored in two bytes, whereby more than one one byte can be used to point to the first occurence because usually less than one byte is required to store the length.\\
|
|
|
|
|
|
-Unfortunally, known implementations like the ones out of LZ Family, do not use probabilities to compress and are therefore out of scope for this work. Since finding repeting sections and their location might also be improved, this chapter will remain.\\
|
|
|
|
|
|
\subsection{Shannons Entropy}
|
|
|
-The founder of information theory Claude Elwood Shannon described entropy and published it in 1948 \cite{Shannon_1948}. In this work he focused on transmitting information. His theorem is applicable to almost any form of communication signal. His findings are not only usefull for forms of information transmition.
|
|
|
+The founder of information theory Claude Elwood Shannon described entropy and published it in 1948 \autocite{Shannon_1948}. In this work he focused on transmitting information. His theorem is applicable to almost any form of communication signal. His findings are not only usefull for forms of information transmition.
|
|
|
|
|
|
% todo insert Fig. 1 shannon_1948
|
|
|
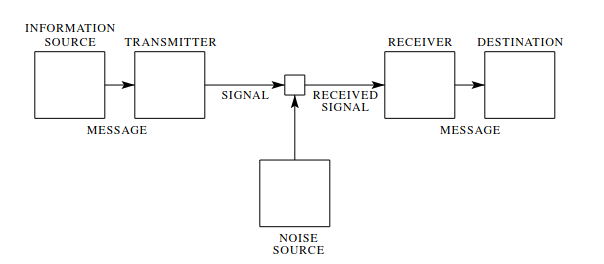
+\begin{figure}[H]
|
|
|
+ \centering
|
|
|
+ \includegraphics[width=15cm]{k4/com-sys.png}
|
|
|
+ \caption{Schematic diagram of a general communication system by Shannons definition. \autocite{Shannon_1948}}
|
|
|
+ \label{k4:comsys}
|
|
|
+\end{figure}
|
|
|
|
|
|
-Altering this figure shows how it can be used for other technology like compression.\\
|
|
|
-The Information source and destination are left unchanged, one has to keep in mind, that it is possible that both are represented by the same phyiscal actor.
|
|
|
-transmitter and receiver are changed to compression/encoding and decompression/decoding and inbetween ther is no signal but any period of time \cite{Shannon_1948}.\\
|
|
|
+Altering \ref{k4:comsys} would show how this can be applied to other technology like compression. The Information source and destination are left unchanged, one has to keep in mind, that it is possible that both are represented by the same phyiscal actor.
|
|
|
+Transmitter and receiver would be changed to compression/encoding and decompression/decoding and inbetween ther is no signal but any period of time \autocite{Shannon_1948}.\\
|
|
|
|
|
|
Shannons Entropy provides a formular to determine the 'uncertainty of a probability distribution' in a finite field.
|
|
|
|
|
|
@@ -83,7 +88,7 @@ Shannons Entropy provides a formular to determine the 'uncertainty of a probabil
|
|
|
% \label{k4:entropy}
|
|
|
%\end{figure}
|
|
|
|
|
|
-He defined entropy as shown in figure \eqref{eq:entropy}. Let X be a finite probability space. Then x in X are possible final states of an probability experimen over X. Every state that actually occours, while executing the experiment generates infromation which is meassured in \textit{Bits} with the part of the formular displayed in \ref{eq:info-in-bits}\cite{delfs_knebl,Shannon_1948}:
|
|
|
+He defined entropy as shown in figure \eqref{eq:entropy}. Let X be a finite probability space. Then x in X are possible final states of an probability experimen over X. Every state that actually occours, while executing the experiment generates infromation which is meassured in \textit{Bits} with the part of the formular displayed in \ref{eq:info-in-bits} \autocite{delfs_knebl,Shannon_1948}:
|
|
|
|
|
|
\begin{equation}\label{eq:info-in-bits}
|
|
|
log_2(\frac{1}{prob(x)}) \equiv - log_2(prob(x)).
|
|
|
@@ -105,7 +110,7 @@ He defined entropy as shown in figure \eqref{eq:entropy}. Let X be a finite prob
|
|
|
\subsection{Arithmetic coding}
|
|
|
This coding method is an approach to solve the problem of wasting memeory due to the overhead which is created by encoding certain lenghts of alphabets in binary. For example: Encoding a three-letter alphabet requires at least two bit per letter. Since there are four possilbe combinations with two bits, one combination is not used, so the full potential is not exhausted. Looking at it from another perspective and thinking a step further: Less storage would be required, if there would be a possibility to encode more than one letter in two bit.\\
|
|
|
Dr. Jorma Rissanen described arithmetic coding in a publication in 1976 \autocite{ris76}. % Besides information theory and math, he also published stuff about dna
|
|
|
-This works goal was to define an algorithm that requires no blocking. Meaning the input text could be encoded as one instead of splitting it and encoding the smaller texts or single symbols. He stated that the coding speed of arithmetic coding is comparable to that of conventional coding methods \cite{ris76}.
|
|
|
+This works goal was to define an algorithm that requires no blocking. Meaning the input text could be encoded as one instead of splitting it and encoding the smaller texts or single symbols. He stated that the coding speed of arithmetic coding is comparable to that of conventional coding methods \autocite{ris76}.
|
|
|
|
|
|
% unusable because equation is only half correct
|
|
|
\mycomment{
|
|
|
@@ -140,19 +145,28 @@ To store as few informations as possible and due to the fact that fractions in b
|
|
|
% its finite subdividing because of the limitation that comes with processor architecture
|
|
|
|
|
|
For the decoding process to work, the \ac{EOF} symbol must be be present as the last symbol in the text. The compressed file will store the probabilies of each alphabet symbol as well as the floatingpoint number. The decoding process executes in a simmilar procedure as the encoding. The stored probabilies determine intervals. Those will get subdivided, by using the encoded floating point as guidance, until the \ac{EOF} symbol is found. By noting in which interval the floating point is found, for every new subdivision, and projecting the probabilies associated with the intervals onto the alphabet, the origin text can be read.\\
|
|
|
-% sclaing
|
|
|
-In computers, arithmetic operations on floating point numbers are processed with integer representations of given floating point number \cite{ieee-float}. The number 0.4 + would be represented by $4\cdot 10^-1$.\\
|
|
|
+% rescaling
|
|
|
+% math and computers
|
|
|
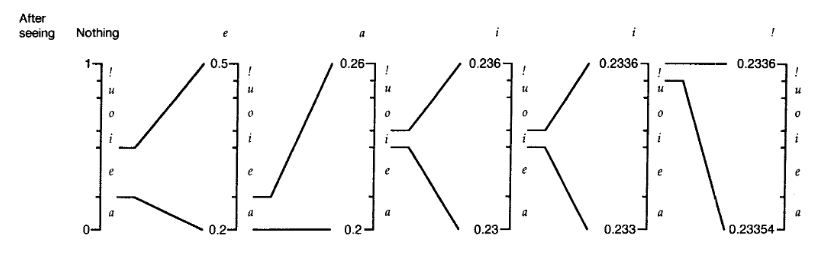
+In computers, arithmetic operations on floating point numbers are processed with integer representations of given floating point number \autocite{ieee-float}. The number 0.4 + would be represented by $4\cdot 10^-1$.\\
|
|
|
Intervals for the first symbol would be represented by natural numbers between 0 and 100 and $... \cdot 10^-x$. \texttt{x} starts with the value 2 and grows as the intgers grow in length, meaning only if a uneven number is divided. For example: Dividing a uneven number like $5\cdot 10^-1$ by two, will result in $25\cdot 10^-2$. On the other hand, subdividing $4\cdot 10^y$ by two, with any negativ real number as y would not result in a greater \texttt{x} the length required to display the result will match the length required to display the input number.\\
|
|
|
+% example
|
|
|
+\begin{figure}[H]
|
|
|
+ \centering
|
|
|
+ \includegraphics[width=15cm]{k4/arith-resize.png}
|
|
|
+ \caption{Illustrative rescaling in arithmetic coding process. \autocite{witten87}}
|
|
|
+ \label{k4:rescale}
|
|
|
+\end{figure}
|
|
|
+
|
|
|
% finite percission
|
|
|
-The described coding is only feasible on machines with infinite percission. As soon as finite precission comes into play, the algorithm must be extendet, so that a certain length in the resulting number will not be exceeded. Since digital datatypes are limited in their capacity, like unsigned 64-bit integers which can store up to $2^64-1$ bits or any number between 0 and 18.446.744.073.709.551.615. That might seem like a great ammount at first, but considering a unfavorable alphabet, that extends the results lenght by one on each symbol that is read, only texts with the length of 63 can be encoded (62 if \acs{EOF} is exclued).
|
|
|
+The described coding is only feasible on machines with infinite percission. As soon as finite precission comes into play, the algorithm must be extendet, so that a certain length in the resulting number will not be exceeded. Since digital datatypes are limited in their capacity, like unsigned 64-bit integers which can store up to $2^64-1$ bits or any number between 0 and 18,446,744,073,709,551,615. That might seem like a great ammount at first, but considering a unfavorable alphabet, that extends the results lenght by one on each symbol that is read, only texts with the length of 63 can be encoded (62 if \acs{EOF} is exclued).
|
|
|
|
|
|
\label{k4:huff}
|
|
|
\subsection{Huffman encoding}
|
|
|
% list of algos and the tools that use them
|
|
|
-D. A. Huffmans work focused on finding a method to encode messages with a minimum of redundance. He referenced a coding procedure developed by Shannon and Fano and named after its developers, which worked similar. The \ac{SF} coding is not used today, due to the superiority in both efficiency and effectivity, in comparison to Huffman. % todo any source to last sentence. Rethink the use of finite in the following text
|
|
|
-Even though his work was released in 1952, the method he developed is in use today. Not only tools for genome compression but in compression tools with a more general ussage \cite{rfcgzip}.\\
|
|
|
-Compression with the Huffman algorithm also provides a solution to the problem, described at the beginning of \ref{k4:arith}, on waste through unused bits, for certain alphabet lengths. Huffman did not save more than one symbol in one bit, like it is done in arithmetic coding, but he decreased the number of bits used per symbol in a message. This is possible by setting individual bit lengths for symbols, used in the text that should get compressed \cite{huf52}.
|
|
|
-As with other codings, a set of symbols must be defined. For any text constructed with symbols from mentioned alphabet, a binary tree is constructed, which will determine how the symbols will be encoded. As in arithmetic coding, the probability of a letter is calculated for given text. The binary tree will be constructed after following guidelines \cite{alok17}:
|
|
|
+D. A. Huffmans work focused on finding a method to encode messages with a minimum of redundance. He referenced a coding procedure developed by Shannon and Fano and named after its developers, which worked similar. The Shannon-Fano coding is not used today, due to the superiority in both efficiency and effectivity, in comparison to Huffman. % todo any source to last sentence. Rethink the use of finite in the following text
|
|
|
+Even though his work was released in 1952, the method he developed is in use today. Not only tools for genome compression but in compression tools with a more general ussage \autocite{rfcgzip}.\\
|
|
|
+Compression with the Huffman algorithm also provides a solution to the problem, described at the beginning of \ref{k4:arith}, on waste through unused bits, for certain alphabet lengths. Huffman did not save more than one symbol in one bit, like it is done in arithmetic coding, but he decreased the number of bits used per symbol in a message. This is possible by setting individual bit lengths for symbols, used in the text that should get compressed \autocite{huf52}.
|
|
|
+As with other codings, a set of symbols must be defined. For any text constructed with symbols from mentioned alphabet, a binary tree is constructed, which will determine how the symbols will be encoded. As in arithmetic coding, the probability of a letter is calculated for given text. The binary tree will be constructed after following guidelines \autocite{alok17}:
|
|
|
% greedy algo?
|
|
|
\begin{itemize}
|
|
|
\item Every symbol of the alphabet is one leaf.
|
|
|
@@ -163,14 +177,14 @@ As with other codings, a set of symbols must be defined. For any text constructe
|
|
|
\end{itemize}
|
|
|
%todo tree building explanation
|
|
|
% storytime might need to be rearranged
|
|
|
-A often mentioned difference between \acs{FA} and Huffman coding, is that first is working top down while the latter is working bottom up. This means the tree starts with the lowest weights. The nodes that are not leafs have no value ascribed to them. They only need their weight, which is defined by the weights of their individual child nodes \cite{moffat20, alok17}.\\
|
|
|
+A often mentioned difference between Shannon-Fano and Huffman coding, is that first is working top down while the latter is working bottom up. This means the tree starts with the lowest weights. The nodes that are not leafs have no value ascribed to them. They only need their weight, which is defined by the weights of their individual child nodes \autocite{moffat20, alok17}.\\
|
|
|
|
|
|
-Given \texttt{K(W,L)} as a node structure, with the weigth or probability as \texttt{$W_{i}$} and codeword length as \texttt{$L_{i}$} for the node \texttt{$K_{i}$}. Then will \texttt{$L_{av}$} be the average length for \texttt{L} in a finite chain of symbols, with a distribution that is mapped onto \texttt{W} \cite{huf}.
|
|
|
+Given \texttt{K(W,L)} as a node structure, with the weigth or probability as \texttt{$W_{i}$} and codeword length as \texttt{$L_{i}$} for the node \texttt{$K_{i}$}. Then will \texttt{$L_{av}$} be the average length for \texttt{L} in a finite chain of symbols, with a distribution that is mapped onto \texttt{W} \autocite{huf}.
|
|
|
\begin{equation}\label{eq:huf}
|
|
|
L_{av}=\sum_{i=0}^{n-1}w_{i}\cdot l_{i}
|
|
|
\end{equation}
|
|
|
The equation \eqref{eq:huf} describes the path, to the desired state, for the tree. The upper bound \texttt{n} is assigned the length of the input text. The touple in any node \texttt{K} consists of a weight \texttt{$w_{i}$}, that also references a symbol, and the length of a codeword \texttt{$l_{i}$}. This codeword will later encode a single symbol from the alphabet. Working with digital codewords, an element in \texttt{l} contains a sequence of zeros and ones. Since there in this coding method, there is no fixed length for codewords, the premise of \texttt{prefix free code} must be adhered to. This means there can be no codeword that match the sequence of any prefix of another codeword. To illustrate this: 0, 10, 11 would be a set of valid codewords but adding a codeword like 01 or 00 would make the set invalid because of the prefix 0, which is already a single codeword.\\
|
|
|
-With all important elements described: the sum that results from this equation is the average length a symbol in the encoded input text will require to be stored \cite{huf52, moffat20}.
|
|
|
+With all important elements described: the sum that results from this equation is the average length a symbol in the encoded input text will require to be stored \autocite{huf52, moffat20}.
|
|
|
|
|
|
% example
|
|
|
% todo illustrate
|
|
|
@@ -185,24 +199,47 @@ With the fact in mind, that left branches are assigned with 0 and right branches
|
|
|
\texttt{A -> 0, C -> 11, T -> 100, G -> 101}.\\
|
|
|
Since high weightened and therefore often occuring leafs are positioned to the left, short paths lead to them and so only few bits are needed to encode them. Following the tree on the other side, the symbols occur more rarely, paths get longer and so do the codeword. Applying \eqref{eq:huf} to this example, results in 1.45 bits per encoded symbol. In this example the text would require over one bit less storage for every second symbol.\\
|
|
|
% impacting the dark ground called reality
|
|
|
-Leaving the theory and entering the practice, brings some details that lessen this improvement by a bit. A few bytes are added through the need of storing the information contained in the tree. Also, like described in % todo add ref to k3 formats
|
|
|
-most formats, used for persisting \acs{DNA}, store more than just nucleotides and therefore require more characters. What compression ratios implementations of huffman coding provide, will be discussed in \ref{k5:results}.\\
|
|
|
+Leaving the theory and entering the practice, brings some details that lessen this improvement by a bit. A few bytes are added through the need of storing the information contained in the tree. Also, like described in \ref{chap:file formats} most formats, used for persisting \acs{DNA}, store more than just nucleotides and therefore require more characters. What compression ratios implementations of huffman coding provide, will be discussed in \ref{k5:results}.\\
|
|
|
|
|
|
-\section{DEFLATE}
|
|
|
+\section{Implementations in Relevant Tools}
|
|
|
+This section should give the reader a quick overview, how a small variety of compression tools implement described compression algorithms.
|
|
|
+
|
|
|
+\subsection{\ac{GeCo}} % geco
|
|
|
+% geco.c: analyze data/open files, parse to determine fileformat, create alphabet
|
|
|
+
|
|
|
+% explain header files
|
|
|
+The header files, that this tool includes in \texttt{geco.c}, can be split into three categories: basic operations, custom operations and compression algorithms.
|
|
|
+The basic operations include header files for general purpose functions, that can be found in almost any c++ Project. The provided functionality includes operations for text-output on the command line inferface, memory management, random number generation and several calculations on up to real numbers.\\
|
|
|
+Custom operations happens to include general purpose functions too, with the difference that they were written, altered or extended by \acs{GeCo}s developer. The last category cosists of several C Files, containing implementations of two arithmetic coding implementations: \textbf{first} \texttt{bitio.c} and \texttt{arith.c}, \textbf{second} \texttt{arith\_aux.c}.\\
|
|
|
+The first two were developed by John Carpinelli, Wayne Salamonsen, Lang Stuiver and Radford Neal (is only mentioned in the latter). Comparing the two files, \texttt{bitio.c} has less code, shorter comments and much more not functioning code sections. Overall the conclusion would be likely that \texttt{arith.c} is some kind of official release, wheras \texttt{bitio.c} severs as a experimental file for the developers to create proof of concepts. The described files adapt code from Armando J. Pinho licenced by University of Aveiro DETI/IEETA written in 1999.\\
|
|
|
+The second implementation was also licensed by University of Aveiro DETI/IEETA, but no author is mentioned. From interpreting the function names and considering the lenght of function bodys \texttt{arith\_aux.c} could serve as a wrapper for basic functions that are often used in arithmetic coding.\\
|
|
|
+Since original versions of the files licensed by University of Aveiro could not be found, there is no way to determine if the files comply with their originals or if changes has been made. This should be considered while following the static analysis.
|
|
|
+
|
|
|
+Following function calls in all three files led to the conclusion that the most important function is defined as \texttt{arithmetic\_encode} in \texttt{arith.c}. In this function the actual artihmetic encoding is executed. This function has no redirects to other files, only one function call \texttt{ENCODE\_RENORMALISE} the remaining code consists of arithmetic operations only.
|
|
|
+% if there is a chance for improvement, this function should be consindered as a entry point to test improving changes.
|
|
|
+
|
|
|
+%useless? -> Both, \texttt{bitio.c} and \texttt{arith.c} are pretty simliar. They were developed by the same authors, execpt for Radford Neal who is only mentioned in \texttt{arith.c}, both are based on the work of A. Moffat \autocite{moffat_arith}.
|
|
|
+%\subsection{genie} % genie
|
|
|
+\subsection{Samtools} % samtools
|
|
|
+\subsubsection{BAM}
|
|
|
+Compression in this fromat is done by a implementation called BGZF, which is a block compression on top of a widely used algorithm called DEFLATE.
|
|
|
+\label{k4:deflate}
|
|
|
+\paragraph{DEFLATE}
|
|
|
% mix of huffman and LZ77
|
|
|
-The DEFLATE compression algorithm combines \ac{LZ77} and huffman coding. It is used in well known tools like gzip.
|
|
|
+The DEFLATE compression algorithm combines \acs{LZ77} and huffman coding. It is used in well known tools like gzip.
|
|
|
Data is split into blocks. Each block stores a header consisting of three bits. A single block can be stored in one of three forms. Each of which is represented by a identifier that is stored with the last two bits in the header.
|
|
|
\begin{itemize}
|
|
|
\item \texttt{00} No compression.
|
|
|
\item \texttt{01} Compressed with a fixed set of Huffman codes.
|
|
|
\item \texttt{10} Compressed with dynamic Huffman codes.
|
|
|
\end{itemize}
|
|
|
-The last combination \texttt{11} is reserved to mark a faulty block. The third, leading bit is set to flag the last data block \cite{rfc1951}.
|
|
|
+The last combination \texttt{11} is reserved to mark a faulty block. The third, leading bit is set to flag the last data block \autocite{rfc1951}.
|
|
|
% lz77 part
|
|
|
-As described in \ref{k4:lz77} a compression with \acs{LZ77} results in literals, a length for each literal and pointers that are represented by the distance between pointer and the literal it points to.\\
|
|
|
-The \acs{LZ77} algorithm is executed before the huffman algorithm. Further compression steps differ from the already described algorithm and will extend to the end of this section.\\
|
|
|
+As described in \ref{k4:lz77} a compression with \acs{LZ77} results in literals, a length for each literal and pointers that are represented by the distance between pointer and the literal it points to.
|
|
|
+The \acs{LZ77} algorithm is executed before the huffman algorithm. Further compression steps differ from the already described algorithm and will extend to the end of this section.
|
|
|
+
|
|
|
% huffman part
|
|
|
-Besides header bits and a data block, two Huffman code trees are store. One encodes literals and lenghts and the other distances. They happen to be in a compact form. This archived by a addition of two rules on top of the rules described in \ref{k4:huff}: Codes of identical lengths are orderd lexicographically, directed by the characters they represent. And the simple rule: shorter codes precede longer codes.\\
|
|
|
+Besides header bits and a data block, two Huffman code trees are store. One encodes literals and lenghts and the other distances. They happen to be in a compact form. This archived by a addition of two rules on top of the rules described in \ref{k4:huff}: Codes of identical lengths are orderd lexicographically, directed by the characters they represent. And the simple rule: shorter codes precede longer codes.
|
|
|
To illustrated this with an example:
|
|
|
For a text consisting out of \texttt{C} and \texttt{G}, following codes would be set for a encoding of two bit per character: \texttt{C}: 00, \texttt{G}: 01. With another character \texttt{A} in the alphabet, which would occour more often than the other two characters, the codes would change to a representation like this:
|
|
|
|
|
|
@@ -220,13 +257,30 @@ For a text consisting out of \texttt{C} and \texttt{G}, following codes would be
|
|
|
\end{footnotesize}
|
|
|
\rmfamily
|
|
|
|
|
|
-Since \texttt{A} precedes \texttt{C} and \texttt{G}, it is represented with a 0. To maintain prefix-free codes, the two remaining codes are not allowed to start with a 0. \texttt{C} precedes \texttt{G} lexicographically, therefor the (in a numerical sense) smaller code is set to represent \texttt{C}.\\
|
|
|
-With this simple rules, the alphabet can be compressed too. Instead of storing codes itself, only the codelength stored \cite{rfc1951}. This might seem unnecessary when looking at a single compressed bulk of data, but when compressing blocks of data, a samller alphabet can make a relevant difference.
|
|
|
+Since \texttt{A} precedes \texttt{C} and \texttt{G}, it is represented with a 0. To maintain prefix-free codes, the two remaining codes are not allowed to start with a 0. \texttt{C} precedes \texttt{G} lexicographically, therefor the (in a numerical sense) smaller code is set to represent \texttt{C}.
|
|
|
+With this simple rules, the alphabet can be compressed too. Instead of storing codes itself, only the codelength stored \autocite{rfc1951}. This might seem unnecessary when looking at a single compressed bulk of data, but when compressing blocks of data, a samller alphabet can make a relevant difference.\\
|
|
|
|
|
|
% example header, alphabet, data block?
|
|
|
+BGZF extends this by creating a series of blocks. Each can not extend a limit of 64 Kilobyte. Each block contains a standard gzip file header, followed by compressed data.\\
|
|
|
|
|
|
-\section{Implementations in Relevant Tools}
|
|
|
-\subsection{} % geco
|
|
|
-\subsection{} % genie
|
|
|
-\subsection{} % samtools
|
|
|
+\subsubsection{CRAM}
|
|
|
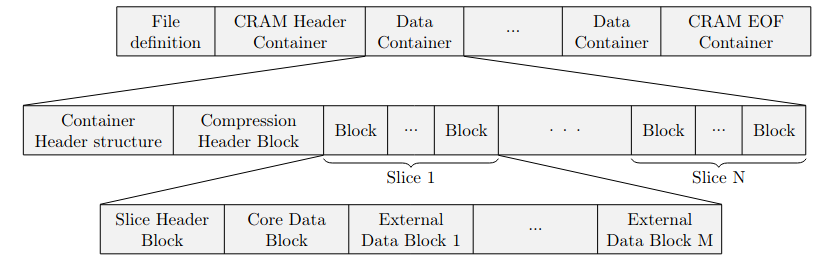
+The improvement of \acs{BAM} \autocite{cram-origin} called \acs{CRAM}, also features a block structure \autocite{bam}. The whole file can be seperated into four sections, stored in ascending order: File definition, a CRAM Header Container, multiple Data Container and a final CRAM EOF Container.\\
|
|
|
+The File definition consists of 26 uncompressed bytes, storing formating information and a identifier. The CRAM header contains meta information about Data Containers and is optionally compressed with gzip. This container can also contain a uncompressed zero-padded section, reseved for \acs{SAM} header information \autocite{bam}. This saves time, in case the compressed file is altered and its compression need to be updated. The last container in a \acs{CRAM} file serves as a indicator that the \acs{EOF} is reached. Since in addition information about the file and its structure is stored, a maximum of 38 uncompressed bytes can be reached.\\
|
|
|
+A Data Container can be split into three sections. From this sections the one storing the actual sequence consists of blocks itself, displayed in \ref FIGURE as the bottom row.
|
|
|
+\begin{itemize}
|
|
|
+ \item Container Header.
|
|
|
+ \item Compression Header.
|
|
|
+ \item A variable amount of Slices.
|
|
|
+ \begin{itemize}
|
|
|
+ \item Slice Header.
|
|
|
+ \item Core Data Block.
|
|
|
+ \item A variable amount of External Data Blocks.
|
|
|
+ \end{itemize}
|
|
|
+\end{itemize}
|
|
|
+The Container Header stores information on how to decompress the data stored in the following block sections. The Compression Header contains information about what kind of data is stored and some encoding information for \acs{SAM} specific flags. The actual data is stored in the Data Blocks. Those consist of encoded bit streams. According to the Samtools specification, the encoding can be one of the following: External, Huffman and two other methods which happen to be either a form of huffman coding or a shortened binary representation of integers. The External option allows to use gzip, bzip2 which is a form of multiple coding methods including run length encoding and huffman, a encoding from the LZ family called LZMA or a combination of arithmetic and huffman coding called rANS.
|
|
|
+% possible encodings:
|
|
|
+% external: no encoding or gzip, bzip2, lzma
|
|
|
+% huffman
|
|
|
+% Byte array coding -> huffman or external...
|
|
|
+% Beta coding -> binary representation
|
|
|
|

 u
u
{kind=link}
{kind=link}
{kind=link}
{kind=link}